QUERY INPUT:
Query Organism: enter organism common name, scientific name or NCBI tax ID. Organisms that are not found in the drop down box were not included in our study.
Query Protein: enter protein names, common or systematic gene names, or protein ID (Ensembl, JGI, Refseq, Uniprot). By clicking "By Fasta', users can also enter the protein sequence. If the protein sequence is not included in ProtPhylo, as for example for newly sequenced organisms, the user can search for an orthologous sequence in a species of interest as the first step. ProtPhylo would then predict functional associations based on the matched protein.
Species WITH the Phenotypic Properties: enter common name, scientific name or NCBI tax ID of organisms with the phenotype of interest. Each organism needs to be entered one at a time.
Species WITHOUT the Phenotypic Properties (Optional): enter common name, scienfific name or NCBI tax ID of organisms without the phenotype of interest. Each organism needs to be entered one at a time.
Orthology Method: users can choose among four established methods to search for orthologs of the query protein (One-way Best Hits, Best Reciprocal Hits, OrthoMCL (1), eggNOG (2)) of the query protein. One-way Best Hits is set as default.
- One-way Best Hits: the best scoring match (E<1e-5) of a query protein in another organism.
- Best Reciprocal Hits: proteins encoded by two genes, each in a different organism, are defined as best reciprocal hits when they find each other as the best scoring match (E<1e-5) in the other organism.
- eggNOG v4 NOG: nonsupervised orthologous groups (NOGs) derived from eggNOG v4.0 (2).
- OrthoMCL: orthologous groups derived from 2048 species using OrthoMCL algorithm (1) with default parameters (BLASTP E<1e-5, 50% match length, 1.5 inflation index). The percent match length refers to the percent positive substitutions from BLASTP; the inflation index is the parameter used by Markov Chain Clustering (MCL) (3) to define the tightness of the clusters. The user can also choose 0% match length and two other inflation indexes (1.1 and 5) to define the orthologous groups.
Click "Search" to predict protein-protein or phenotype-to-protein functional associations based on the above criteria. The list of candidate proteins can be further prioritized by applying any combination of filter options (listed below). Alternatively, filter options can be selected before the run to obtain a pre-filtered ranked list of candidates predicted to be functional associated with the query protein or phenotype.
FILTER OPTIONS:
Profile Similarity:
- HD (Hamming Distance): the Hamming distance between the phylogenetic profiles of two proteins represents the number of organisms that do not have the same presence/absence of orthologs (4). The smaller the HD, the more similar the evolutionary history. For phenotype phylogenetic profiling, the HD is set to zero by default. In this case, ProtPhylo will output a list of candidate proteins that have an ortholog in all of the input organisms with the phenotypic property but lack an ortholog in all of the input organism without the phenotypic property. Users can retrieve proteins with a higher HD to the phenotype of interest by changing the default value.
- HD Percentile: percentage of proteins within a query organism that have the lowest Hamming distance. The 5th percentile is set as default.
Subcellular Localization (Filter Type: AND, OR): users can filter candidate proteins based on common evidence of subcellular localization from more than one prediction method (AND). Alternatively, users can filter candidate proteins based on combined evidence of subcellular localization from any of the selected methods (OR).
- TargetP 1.1: prediction of the subcellular localization of eukaryotic proteins by TargetP 1.1 program (5).
- MitoProt II Score: prediction of mitochondrial targeting sequences by MitoProtII (6).
- LocTree3: prediction of the subcellular localization by LocTree3 (7).
- Mitocarta: prediction of mitochondrial locaized proteins. This is only available for Mouse and Human (8).
- Uniprot: annotation of subcellular localization from UniprotKB. The users can input any text based on the controlled vocabulary for subcellular localization provided by Uniprot (http://www.uniprot.org/docs/subcell).
Others:
- Transmembrane: prediction of transmembrane helices in proteins by TMHMM v2.0 (9) as well as annotated from Uniprot database based on experimental and computational evidence.
- Pfam: prediction of protein functional domains (10) .
- Keyword: any text associated with protein/gene name and ID.
- STRING score: confidence score for each functional association predicted by STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) (11).
QUERY OUTPUT:
The output of a ProtPhylo run is a list of candidate proteins (rows) that are predicted to be functionally associated with a query protein (Protein Phylogenetic Profiling) or phenotype (Phenotype Phylogenetic Profiling) solely based on evidence of coevolution. Candidate proteins are automatically ranked based on their phylogenetic distance (Hamming distance, HD) to the query protein or the phenotype. The lowest the HD the strongest the functional association. The total number of hits (predicted functional associations) is indicated at the top of the list. By default, only the first 100 hits are shown; to display the next 100 hits, the user can click on the button “Show more results” found on the top right corner below the filter options. Note that the first listed protein of a “Protein Phylogenetic Profiling” output list (highlighted in red) is always the query protein. If all the hits are displayed, the user can sort the hits based on any header. For each protein the following additional information (columns) can be retrieved:
- Source Protein ID: protein ID used in our database. Click the hyperlink to open a new page containing the detailed protein information.
- HD: Hamming distance to the query protein.
- HD Percentile: value indicating the position of the hit within the distribution of HD percentile values for all proteins in the query organism. The HD percentile represents the percentage of proteins within the query organism that have the lowest Hamming distance to the query protein. Note that this value should not be over-interpreted as probability or confidence score.
- Reciprocal HD Percentile: HD percentile of the query protein if the hit is used as the query. To calculate this value, the user needs to click on the magnifier icon.
- Orthologs: name of the orthologous group. By clicking the hyperlink, users can retrieve detailed information on all the proteins that have been identified as orthologs of the query protein based on the selected orthology method. Data can be exported as a csv file.
- Gene Symbol: official gene name as in UniProtKB. Saccharomyces Genome Database, Wormbase, Ensembl are used for the annotation of S. cerevisiae, C. elegans, D. rerio, respectively.
- Uniprot ID: the UniProtKB accession number.
- Uniprot Entry: the UniProtKB entry name.
- Protein Name: the description used by UniProtKB. Saccharomyces Genome Database, Wormbase, Ensembl are used for the annotation of S. cerevisiae, C. elegans, D. rerio respectively.
Click “Export” to export the results into a csv text file.
Protein Phylogenetic Profiling: the MICU1 case study
INPUT:
Users should use the “Protein Phylogenetic Profiling” option to search for additional proteins that could be functionally associated (members of the same protein complex, pathways, receptors and ligands, etc.) to a protein of interest (‘Query Protein’) within a user-defined organism (‘Query Organism’). As an example, we show how ProtPhylo can be used to identify human proteins that are functionally associated to Micu1, the mitochondrial calcium uptake 1 protein (12,13). Micu1 was identified by phenotype phylogenetic profiling in 2010 as the founding member of the human mitochondrial calcium uniporter (12). The latter was then shown to include additional subunits such as Mcu, Mcub, Micu2, and Micu3 (14-16). Can ProtPhylo predict a functional association between Micu1 and other subunits of the mitochondrial calcium uniporter solely based on phylogenetic profiling analysis? First, type and select ‘Homo sapiens’ in the ‘Query Organism’ text box. Next, enter the protein name ‘Micu1’ in the ‘Query Protein’ text box and select ‘One-way Best Hits’ (default) as ‘Orthology Method’. You can select an HD percentile <1st if you only want to retrieve functional associations to the top 1% of human proteins with the lowest Hamming distance to the query protein. Now, click the ‘Search’ button.
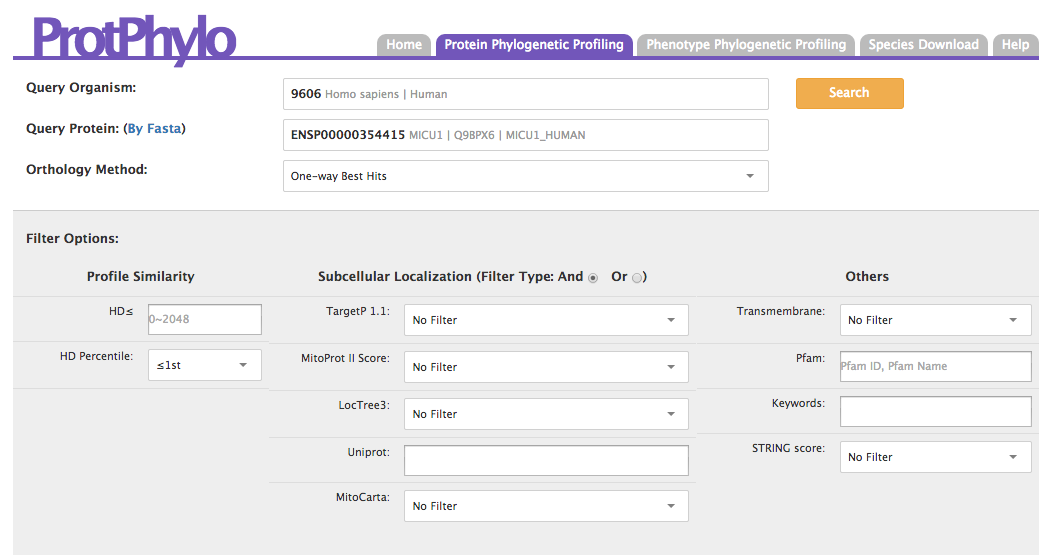
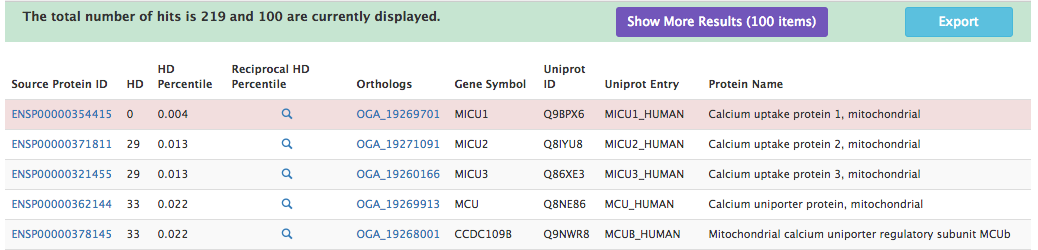
OUTPUT:
ProtPhylo predicts a total of 219 proteins out of all human proteins that match the above criteria. Only the top four hits (lowest HD) are shown below and they include Micu2, Micu3, Mcu, and Mcub, known components of the mitochondrial calcium uniporter. An HD of 29 (Micu2, Micu3) means that the phylogenetic profiles of Micu1, query protein, and Micu2 or Micu3 are identical except for 29 out of 2048 organisms used in the analysis.
Phenotype Phylogenetic Profiling: the MCU case study
INPUT:
Users should use the “Phenotype Phylogenetic Profiling” option when none of the proteins involved in a phenotype of interest are known. Here, users can search for proteins within a user-defined organism (‘Query Organism’) that are functionally associated to a phenotype of interest. An example is shown below for the identification of human mitochondrial proteins involved in mitochondrial calcium uptake (12,17). First, users need to select a query organism for which the phenotype-to-protein associations should be predicted. In the example below, we selected ‘Homo sapiens’ as we are interested in identifying human proteins involved in mitochondrial calcium uptake. Second, in the textbox ‘Species WITH the Phenotype of Interest’, users need to type the common or scientific name of species that show the phenotype of interest. The species need to be entered one at a time. As a result, a string of blue boxes with numbers will appear. The number in each box represents the NCBI tax ID of the selected species. If the phenotype of interest is not conserved in other species, users should enter these species in the textbox ‘Species WITHOUT the Phenotype of Interest’. For example, it is known that mitochondria of yeast cannot uptake calcium, therefore, we enter S. cerevisiae in the textbox ‘Species WITHOUT the Phenotype of Interest’.
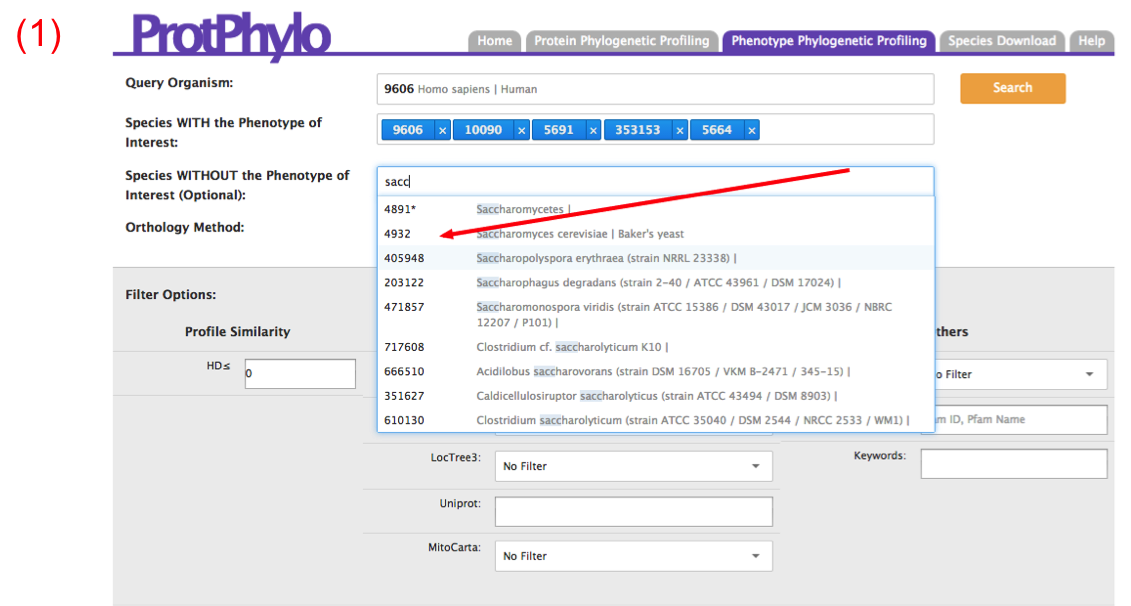
Next, users need to select the ‘Orthology Method’ of choice. Here, we select ‘One-way Best Hits’ as ‘Orthology Method’. As we search for the proteins involved in calcium uptake in mitochondria, we will restrict the analysis to all human proteins that are predicted to be localized in mitochondria based on common (AND) evidence of mitochondrial localization from TargetP, MitoProtII, LocTree and Uniprot. Hamming distance of zero is set as default, therefore only perfect matches will be retrieved. Now, click the ‘Search’ button.
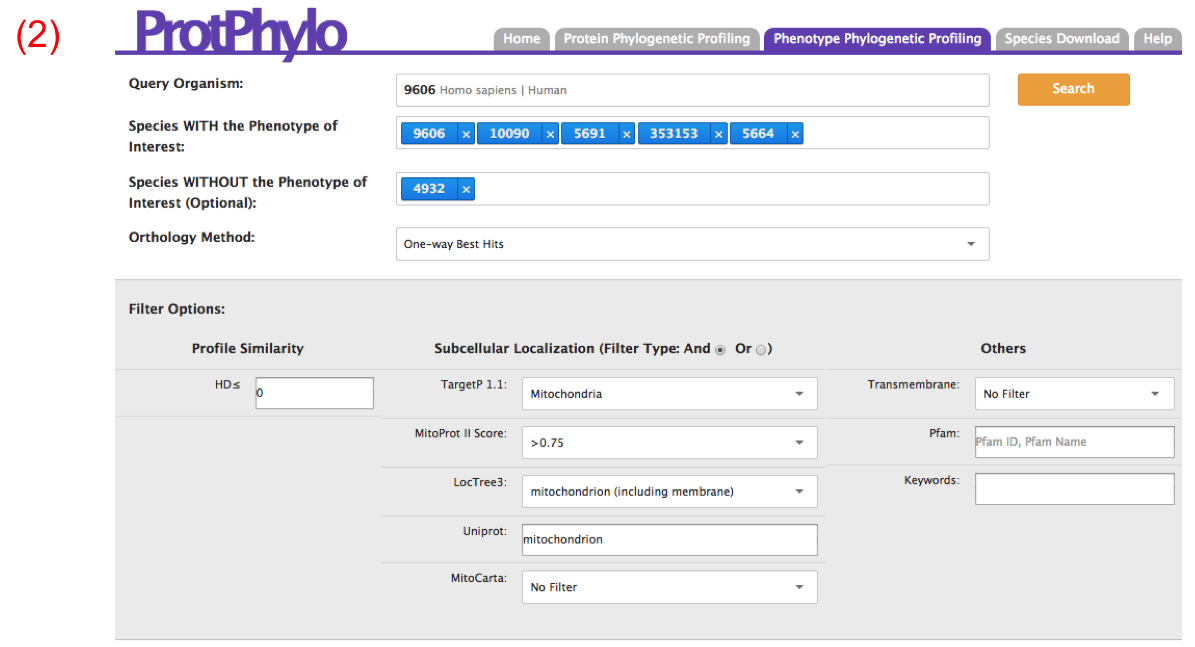
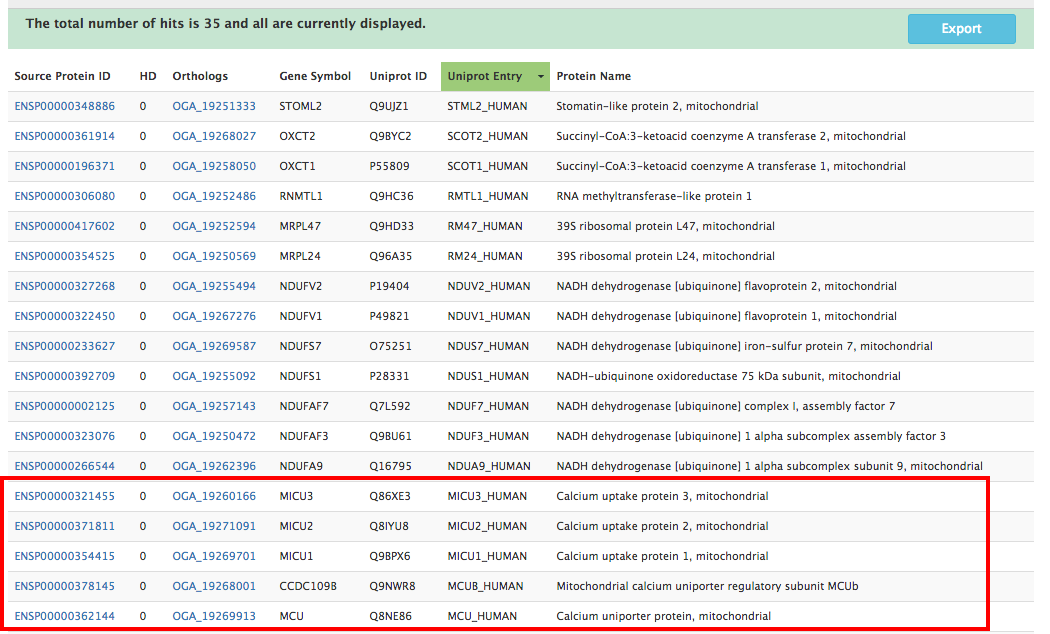
OUTPUT:
ProtPhylo predicts a total of 35 proteins out of all human proteins that match the above criteria. All of the candidate proteins have the same probability to be associated with the phenotype of interest, as all have an Hamming distance of zero. Users can now plan targeted experiments to test whether any of these candidate proteins affect mitochondrial calcium uptake (phenotype of interest). As we can see, the set of candidate proteins include known components of the mitochondrial calcium uniporter (Micu1, Micu2, Micu3, Mcu, and Mcub).
Note: We suggest users to run ProtPhylo with each orthology method first and then evaluate the ranking of known interacting/funcionally associated proteins (if available) based on HD, HD percentile, and the reciprocal HD percentile. We also suggest users, who are not familiar with orthology inference methods, to consult relevant reviews on this topic that provide examples of decision tree for choosing the appropriate orthology detection tool, for example, Kuzniar et al. (18),
REFERENCES:
- Li L., Christian J. Stoeckert, Jr., and David S. Roos (2003). OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 13: 2178-2189.
- Powell S., Forslund K., Szklarczyk D., Trachana K., Roth A., Huerta-Cepas J., et al. . (2014). eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 42, D231–D239.
- Van Dongen, S. (2000). “Graph clustering by flow simulation.” Ph.D thesis, University of Utrecht, The Netherlands.
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO (1999) Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci U S A 96: 4285–4288
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007). Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2:953–971.
- M.G. Claros, P. Vincens (1996). Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241, 779-786.
- Goldberg T., Hecht M., Hamp T., Karl T., Yachdav G., Ahmed N., Altermann U., Angerer P., Ansorge S., Balasz K (2014). LocTree3 prediction of localization. Nucleic Acids Res. 42 (Web Server issue):W350-5
- Pagliarini, D.J., Calvo, S.E., Chang, B., Sheth, S.A., Vafai, S.B., Ong, S.E., Walford, G.A., Sugiana, C., Boneh, A., Chen, W.K., et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112-123.
- A. Krogh, B. Larsson, G. von Heijne, and E. L. L. Sonnhammer (2001). Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes.
Journal of Molecular Biology, 305(3):567-580.
- Finn, R.D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R.Y., Eddy, S.R., Heger, A., Hetherington, K., Holm, L., Mistry, J. et al.
(2014) Pfam: the protein families database. Nucleic acids research, 42, D222-230.
- Franceschini, A., Szklarczyk, D., Frankild, S., Kuhn, M., Simonovic, M., Roth, A., Lin, J., Minguez, P., Bork, P., von Mering, C. et al. (2013) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic acids research, 41, D808-815.
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK (2010). MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 467(7313):291-6.
- Baughman JM*, Perocchi F*, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 476(7360):341-5.
- Plovanich, M., Bogorad, R.L., Sancak, Y., Kamer, K.J., Strittmatter, L., Li, A.A., Girgis, H.S., Kuchimanchi, S., De Groot, J., Speciner, L. et al. (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PloS one, 8, e55785
- Raffaello, A., De Stefani, D., Sabbadin, D., Teardo, E., Merli, G., Picard, A., Checchetto, V., Moro, S., Szabo, I. and Rizzuto, R. (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. The EMBO journal, 32, 2362-2376
- Patron, M., Checchetto, V., Raffaello, A., Teardo, E., Vecellio Reane, D., Mantoan, M., Granatiero, V., Szabo, I., De Stefani, D. and Rizzuto, R. (2014) MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Molecular cell, 53, 726-737
- De Stefani, D., Raffaello, A., Teardo, E., Szabo, I. and Rizzuto, R. (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature, 476, 336-34
- Kuzniar, A., van Ham, R.C., Pongor, S. and Leunissen, J.A. (2008) The quest for orthologs: finding the corresponding gene across genomes. Trends in genetics : TIG, 24, 539-551

